
Research & Development
Our proprietary drug delivery platform provides a unique and beneficial delayed-release and extended-release delivery to a specifically targeted area in the gastrointestinal (GI) tract to ensure medication is active during the most critical times of the day or night.
The drug delivery system, which has been used in our FDA-approved product, has been specifically engineered to optimize the delivery of small or large molecule active ingredients with a wide variety of physiochemical properties. The system has the potential for use in multiple therapeutic areas, including, but not limited to:
- Central nervous system (CNS) disorders
- GI disorders
- Oncology
- Diabetes
- Genetic Therapies
How Does The Drug Delivery Platform Work?
With its unique ability to deliver 100% of the drug payload to the distal GI without the variability associated with GI transit time, pH, and microbiota, the drug delivery platform provides a consistent timing of drug release. The platform’s extended-release phase is customizable based on the delivery needs and can be adjusted to be just a few hours or even days.
About The Capsule
Each capsule contains hundreds of uniform beads with a diameter of ~850 microns.

About The Beads
The drug delivery platform capsules contain a single population of beads, each bead having identical release properties, as opposed to combining beads with varying release properties to achieve the desired pharmacokinetic profile.
A bead is comprised of:
- An outer delayed-release (DR) layer composed of polymers with hydrophobic, hygroscopic, and pH-dependent properties, which contribute to a unique “push-pull” mechanism.
- An inner extended-release (ER) layer composed of hydrophobic and soluble polymers. This layer provides a highly tunable, or flexible, delay period before drug release. Drug release is precisely controlled by a balance of Active Pharmaceutical Ingredient (API) solubility and dynamic film properties.
- A drug-loaded immediate-release (IR) core that acts as a drug reservoir that can contain single or multiple active ingredients, prodrugs, stabilizers, solubility aids, permeation enhancers, etc.
Moving Through The System
While release can be fine-tuned to occur over a period of greater than 36 hours, here is an example of how the drug delivery platform moves through the system in a shorter release:
1
Ingestion → Stomach: After ingestion, the capsule itself dissolves within 15 minutes, releasing hundreds of beads into the gastric contents of the stomach.
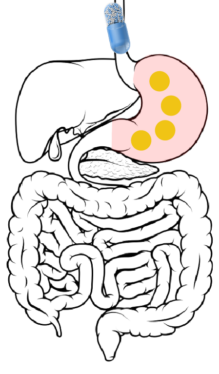
2
Stomach → Duodenum → Jejunum: 2 to 7 hours after ingestion, the beads are released through the pylorus into the duodenum. The DR layer begins wetting but remains insoluble in the upper GI tract.
3
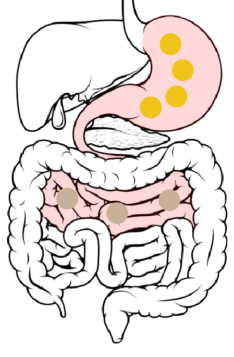
Jejunum → Ileum: 6 to 8 hours after ingestion, the beads spread throughout the small intestine and pass from the jejunum to the ileum.
The DR layer starts to erode, increasing access of GI fluids to the ER layer. The ER layer resists wetting, delaying its dissolution and penetration of GI fluids to the drug-loaded core.
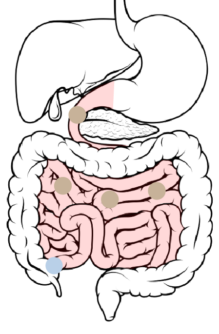
4
Ileum → Cecum: 8-11 hours after ingestion, the beads pass through the ileocecal valve into the cecum.
With increased wetting, the soluble polymer begins to dissolve and the ER layer slowly starts to become permeable to GI fluids. This allows the drug to slowly diffuse into the intestinal lumen.
The hydrophobic polymer repels fluids. This affects the rate at which GI fluids gain accessibility to the drug-loaded core. Dissolution of the soluble polymer results in increased permeability to the drug-loaded core.
This controls the rate at which GI fluids gain accessibility to the drug-loaded core, as well as the rate at which the drug diffuses out of the bead.
5
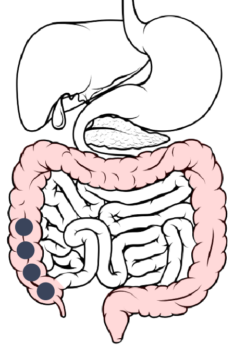
Cecum → Ascending Colon: About 14 hours after ingestion, the ER layer reaches its maximal permeability, resulting in a maximal rate of dissolution and drug absorption.
6
Ascending colon → Transverse colon: The rate of drug dissolution starts to decrease due to a lower concentration gradient.
7
Transverse colon → Descending colon: About 20 hours after ingestion, drug dissolution is complete, however, absorption of the drug continues.
A Successful Use Of The Drug Delivery Platform
Our FDA-approved product is a CNS stimulant prescription medicine used for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) in people 6 years of age and older.
You are about to leave the Ironshore website. If you would like to visit jornaypm.com please select OK.
Have a question, contact us today.

© 2024 Ironshore Pharmaceuticals, Inc. | PRO-CORP-1234-v2 03/24 | Site Design: Digital Elevator